
FDA 524B Is Here: What Medical Device Makers Must Test Now
Contributors


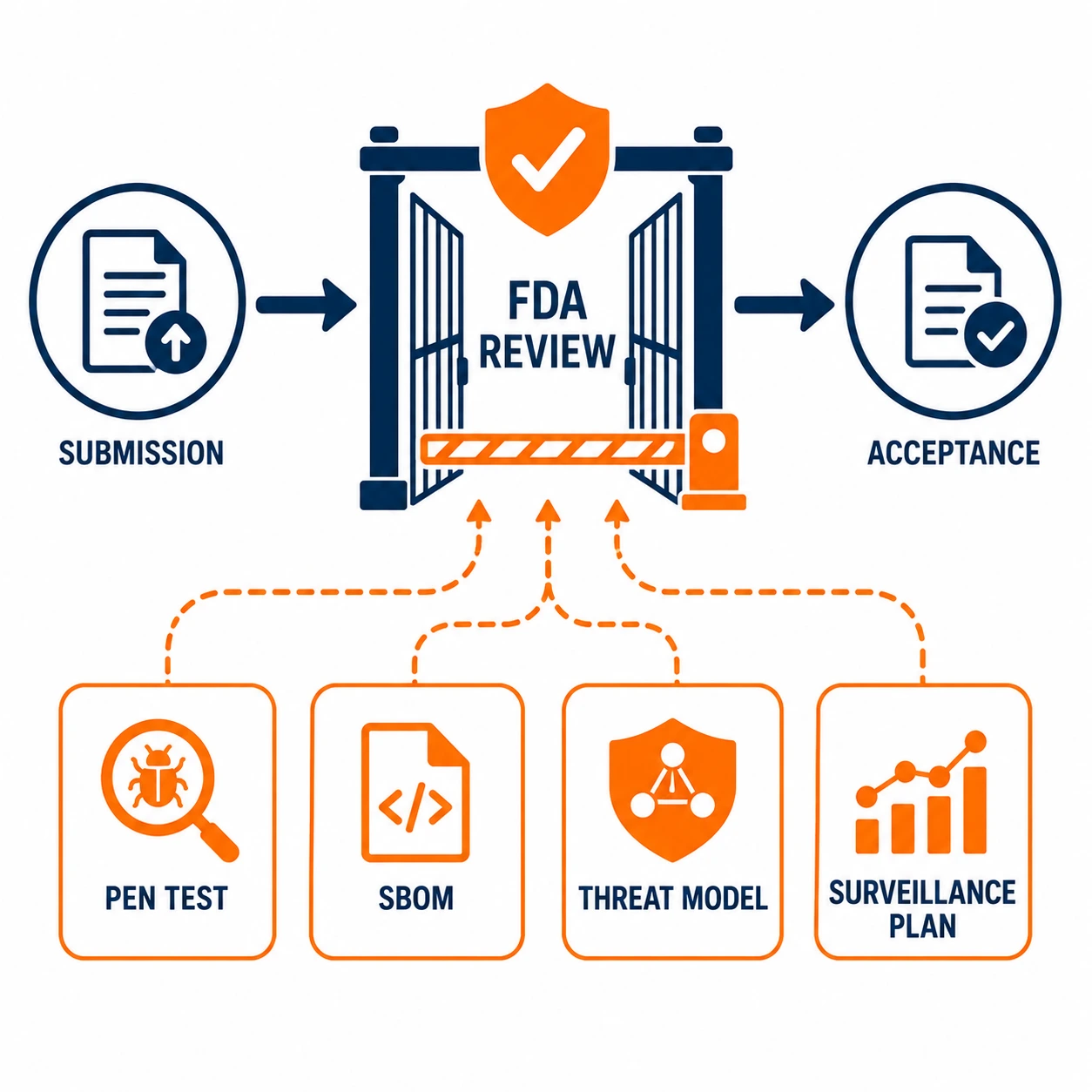
Section 524B of the Federal Food, Drug, and Cosmetic Act changed the regulatory landscape for medical device cybersecurity in a way that guidance documents never did. Guidance is advisory. 524B is law. As of March 2023, the FDA has the authority to refuse to accept premarket submissions for cyber devices that do not include adequate cybersecurity documentation. Understanding what that means for your pen testing program is not optional.
What 524B Actually Requires
Section 524B requires manufacturers of cyber devices to submit cybersecurity information as part of premarket submissions, establish processes to monitor and address postmarket cybersecurity vulnerabilities, and provide a software bill of materials covering commercial, open-source and off-the-shelf components. Penetration testing is a core component of the premarket cybersecurity evidence the FDA expects to see.
The FDA's Refuse to Accept policy means that submissions without adequate cybersecurity sections are rejected before substantive review begins. That is a significant shift from the prior environment, where cybersecurity deficiencies typically surfaced as questions during review rather than grounds for outright rejection at intake. The bar for what constitutes adequate has been raised, and pen testing is part of meeting it.
Which Devices Are Covered
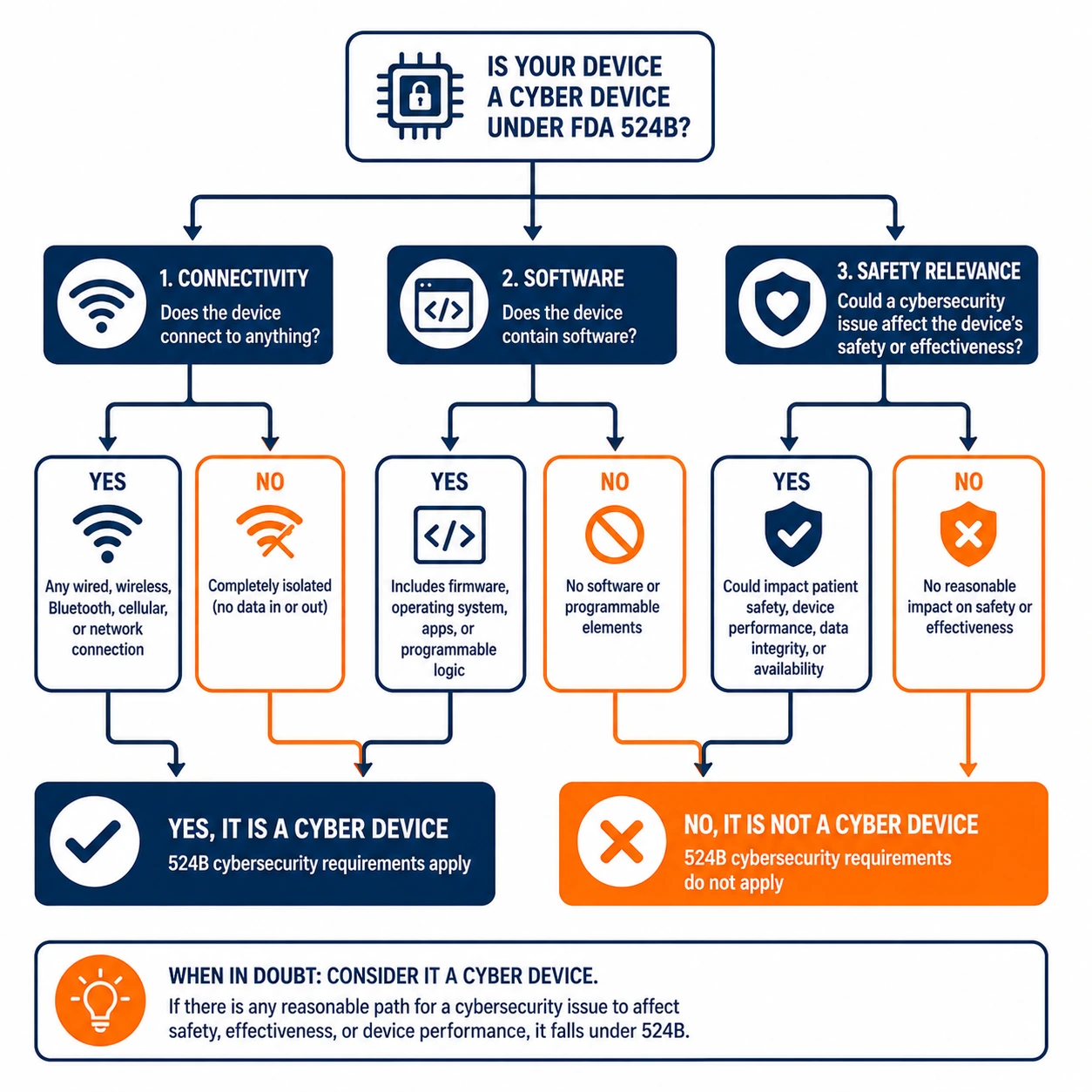
A cyber device under 524B is any device that includes software validated, installed or authorized by the sponsor, has the ability to connect to the internet, and contains any such technological characteristics that could be vulnerable to cybersecurity threats. That definition covers a wide range of devices including infusion pumps, patient monitors, imaging systems and any device with network connectivity or software that affects safety or effectiveness.
Manufacturers should not assume that limited connectivity excludes a device from 524B scope. Bluetooth-only devices, devices that connect only to a proprietary network and devices with indirect internet connectivity through a gateway have all been treated as cyber devices in FDA review contexts. When in doubt, the safer assumption is that 524B applies.
How to Scope a Pen Test for FDA Premarket Submission
The scope of a 524B pen test must reflect the device's intended use environment and its actual attack surface. That means testing hardware interfaces including USB ports, debug interfaces and physical access points. It means testing software and firmware for known vulnerabilities, authentication weaknesses and logic flaws. It means testing communication interfaces including wireless protocols, Bluetooth implementations and any network connectivity the device supports.
The intended use environment matters because it shapes the threat model. A device used in an ICU connected to a hospital network faces a different threat profile than a home-use device connecting to a consumer smartphone app. The pen test methodology should be calibrated to the environment where the device will actually operate, not a generic enterprise IT environment.
What the FDA Looks for in Pen Test Evidence
FDA reviewers evaluating the cybersecurity section of a premarket submission are looking for evidence that the manufacturer understands the device's attack surface, tested against a threat model that reflects real-world adversary behavior, found and addressed vulnerabilities before submission, and has a plan to manage vulnerabilities discovered after clearance.
The pen test report needs to demonstrate all four of these things, not just document what the testers did. That means the report must connect to the threat model, reference the SBOM for context on tested components, document findings with enough detail to show they were genuinely assessed and not just noted, and map to the design controls and risk management file that FDA reviewers will cross-reference.
Postmarket Obligations
524B does not end at clearance. Manufacturers are required to establish processes to monitor for cybersecurity vulnerabilities and to address them in a reasonable time. The FDA expects a patch management plan as part of the premarket submission, and it expects manufacturers to demonstrate that they have the organizational capability to execute on it postmarket.
Building that capability requires more than writing a policy. It requires a tested process for receiving vulnerability disclosures, assessing their relevance to specific device models, developing and validating patches, and communicating with customers and the FDA when significant vulnerabilities are addressed. Pen testing plays a role in validating that process as well as in the initial premarket submission.

Frequently Asked Questions
Section 524B requires makers of cyber devices to do three things: submit cybersecurity information as part of their premarket submission, establish processes to monitor and address postmarket cybersecurity vulnerabilities, and provide a software bill of materials covering commercial, open-source, and off-the-shelf components. Penetration testing is a core part of the premarket cybersecurity evidence the FDA expects to see supporting these requirements.
The difference is legal force. FDA guidance documents are advisory, meaning they describe expectations without binding authority. Section 524B is law, part of the Federal Food, Drug, and Cosmetic Act. Since March 2023, the FDA has had the authority to refuse to accept premarket submissions for cyber devices that do not include adequate cybersecurity documentation, which turns what used to be a recommendation into a condition of submission.
The Refuse to Accept policy means the FDA can reject a premarket submission at intake, before substantive review even begins, if its cybersecurity section is inadequate. This is a significant shift from the prior environment, where cybersecurity gaps surfaced as questions during review rather than as grounds for outright rejection. Adequate penetration testing evidence is part of clearing that intake bar.
A cyber device is any device that includes software validated, installed, or authorized by the sponsor, has the ability to connect to the internet, and contains technological characteristics that could be vulnerable to cybersecurity threats. That definition covers a wide range of products, including infusion pumps, patient monitors, imaging systems, and any device with network connectivity or software that affects safety or effectiveness.
Manufacturers should not assume limited connectivity puts a device outside 524B scope. Bluetooth-only devices, devices that connect only to a proprietary network, and devices with indirect internet connectivity through a gateway have all been treated as cyber devices in FDA review contexts. When connectivity is in question, the safer assumption is that 524B applies and the device needs cybersecurity evidence.
The scope must reflect the device's intended use environment and its actual attack surface. That means testing hardware interfaces such as USB ports, debug interfaces, and physical access points; testing software and firmware for known vulnerabilities, authentication weaknesses, and logic flaws; and testing communication interfaces such as wireless protocols, Bluetooth implementations, and any network connectivity the device supports. The methodology should match where the device actually operates, not a generic enterprise IT environment.
Because the environment shapes the threat model. A device used in an ICU and connected to a hospital network faces a different threat profile than a home-use device that connects to a consumer smartphone app. Calibrating the pen test to the real operating environment, rather than a generic setup, is what makes the evidence credible to FDA reviewers assessing whether the testing reflects realistic adversary behavior.
FDA reviewers look for evidence of four things: that the manufacturer understands the device's attack surface, tested against a threat model reflecting real-world adversary behavior, found and addressed vulnerabilities before submission, and has a plan to manage vulnerabilities discovered after clearance. The report must demonstrate all four, connecting to the threat model, referencing the SBOM, documenting findings in genuine detail, and mapping to the design controls and risk management file.
524B obligations do not end at clearance. Manufacturers must establish processes to monitor for cybersecurity vulnerabilities and address them within a reasonable time, and the FDA expects a patch management plan as part of the premarket submission. It also expects proof of the organizational capability to execute that plan, including a tested process for receiving disclosures, assessing them per device model, validating patches, and communicating with customers and the FDA.
The software bill of materials is a required element of a 524B submission and must cover commercial, open-source, and off-the-shelf components. Beyond satisfying the requirement itself, the SBOM gives context for the penetration test: the pen test report should reference it so reviewers can see which components were tested and understand the device's software supply chain when assessing findings.



Other Popular Articles
In the digital age, businesses must adopt an ad
GRC is the capability, or integrated collection
Get the latest insights straight from our desk to your inbox.
Other Featured Articles
Explore More
What Is CMMC and Do You Need It?
Learn what CMMC is, who needs it, and how to determine if your business requires Level 1 compliance for DoD contracts.
Shantanoo Govilkar
SVP Strategic Solutions Risk & Cybersecurity Solutions view
CMMC Level 1 vs Level 2, Which Applies to You?
Learn the key differences between CMMC Level 1 and Level 2, understand FCI vs. CUI, and determine which CMMC level your DoD contract requires to avoid unnecessary costs or compliance risks.
Shantanoo Govilkar
SVP Strategic Solutions Risk & Cybersecurity Solutions view
What CMMC Means for DoD Subcontractors
Understand what CMMC means for DoD subcontractors, how compliance requirements flow down from prime contractors, who must comply, and the steps to determine your CMMC obligations.
Shantanoo Govilkar
SVP Strategic Solutions Risk & Cybersecurity Solutions view
How to Complete Your CMMC Level 1 Self Assessment
Complete CMMC Level 1 self-assessment guide: define scope, work through 15 practices, fix common gaps, and submit your SPRS affirmation.
Shantanoo Govilkar
SVP Strategic Solutions Risk & Cybersecurity Solutions view
The October 2026 CMMC Deadline: What Happens If You Miss It
Prepare for the October 2026 CMMC deadline. Learn how compliance affects DoD contract eligibility, SPRS affirmations, renewals, and subcontractors.
Shantanoo Govilkar
SVP Strategic Solutions Risk & Cybersecurity Solutions view
Operational Technology (OT) Penetration Testing Guide for Ransomware Defense
Ransomware doesn't need to breach your control room it needs to get close enough that you can't trust it hasn't.
Shantanoo Govilkar
SVP Strategic Solutions Risk & Cybersecurity Solutions view
A Medical Device Maker's Guide to FDA Cybersecurity Testing for 510(k) & PMA
The FDA doesn't publish a pen testing checklist, but its guidance, 524B requirements, and reviewer expectations add up to one.
Shantanoo Govilkar
SVP Strategic Solutions Risk & Cybersecurity Solutions view
Pharma Pen Testing: Why FDA and IP Risk Need Different Scoping
Standard pen test scoping frameworks weren't built for pharma.
Shantanoo Govilkar
SVP Strategic Solutions Risk & Cybersecurity Solutions view
FDA 524B Is Here: What Medical Device Makers Must Test Now
Section 524B made medical device cybersecurity a legal requirement, not a guideline.
Shantanoo Govilkar
SVP Strategic Solutions Risk & Cybersecurity Solutions view
Why CHIPS Act Manufacturers Can't Rely on CMMC Pen Testing Alone
Semiconductor manufacturers face dual compliance obligations under CMMC 2.0 and the CHIPS Act and a standard pen test satisfies neither fully.
Shantanoo Govilkar
SVP Strategic Solutions Risk & Cybersecurity Solutions view
Why Pen Test Evidence Fails C3PAO Assessments (and How to Fix It)
Completing a pen test isn't enough for CMMC.
Shantanoo Govilkar
SVP Strategic Solutions Risk & Cybersecurity Solutions view
PTaaS vs. Annual Pen Testing: Why Manufacturers Are Switching
Annual penetration testing produces documentation, not security.
Shantanoo Govilkar
SVP Strategic Solutions Risk & Cybersecurity Solutions view
Map Your OT Attack Surface Before the Next Audit
Don't wait for an auditor to tell you what you missed.
Shantanoo Govilkar
SVP Strategic Solutions Risk & Cybersecurity Solutions view
How to Scope IT-OT Penetration Testing Safely
Learn how to safely scope IT-OT penetration testing engagements.
Shantanoo Govilkar
SVP Strategic Solutions Risk & Cybersecurity Solutions view
How Often Should Manufacturers Run OT Penetration Testing?
Annual pen testing fits a budget cycle but it doesn't reflect how fast manufacturing environments actually change.
Shantanoo Govilkar
SVP Strategic Solutions Risk & Cybersecurity Solutions view
Pharmaceutical Pen Testing: What 21 CFR Part 11 and cGMP Require
21 CFR Part 11 and cGMP don't mention penetration testing but the controls they require depend on it.
Shantanoo Govilkar
SVP Strategic Solutions Risk & Cybersecurity Solutions view
Pharmaceutical Pen Testing: Why R&D and GxP Need Different Scopes
R&D and GxP regulated environments have different risk profiles, compliance requirements, and testing constraints.
Shantanoo Govilkar
SVP Strategic Solutions Risk & Cybersecurity Solutions view
Why Pharmaceutical Pen Testing Must Address Nation-State Threats
Nation-state actors treat pharma like critical infrastructure targeting formulation data, synthesis routes, and clinical IP with patience and precision.
Shantanoo Govilkar
SVP Strategic Solutions Risk & Cybersecurity Solutions view
How Ransomware Crosses the IT-OT Boundary (And How to Stop It)
Ransomware operators target the IT-OT boundary deliberately and they know manufacturing economics well.
Shantanoo Govilkar
SVP Strategic Solutions Risk & Cybersecurity Solutions view
Where Industry 4.0 Left Your OT Attack Surface Wide Open
Industry 4.0 connected OT environments were never built for. Learn why traditional IT security tools fall short and what OT penetration testing reveals that audits miss.
Shantanoo Govilkar
SVP Strategic Solutions Risk & Cybersecurity Solutions view
What AS4 Actually Solves: Real Outcomes Companies See After Migration
Discover what AS4 actually solves for modern businesses. Learn the real outcomes companies achieve after migration, from stronger security to better B2B integration performance.
EDI Solutions Group
Marketing Group view
7 Migration Pitfalls That Derail AS4 Upgrades (and How to Avoid Them)
Avoid costly AS4 upgrade mistakes. Discover 7 migration pitfalls that delay projects, create risk, and disrupt B2B messaging, plus practical ways to avoid them.
EDI Solutions Group
Marketing Group view
How to Perform Penetration Testing in Cloud Environments (AWS, Azure, and GCP) - 2026 Edition
A practical guide to cloud penetration testing across AWS, Azure, and GCP. Learn methods, tools, and best practices to identify vulnerabilities and improve security.
Cybersecurity Solutions Group
Marketing Group view
5 Signs It's Time to Move Legacy EDI Environment to AS4 Protocol
Partner onboarding delays, compliance gaps, and rising maintenance costs are signals your EDI infrastructure is reaching its limits. Learn the five signs it is time to evaluate a move to AS4.
EDI Solutions Group
Marketing Group view
How to Design Custom Chatbots That Cannot “Make Stuff Up”
Confident AI answers without traceable sources create institutional risk. Learn how Grounded RAG architecture retrieves real documents first and attaches verifiable citations to every response.
Data and AI Solutions Group
Marketing Group view
How Citation-Backed Conversational AI Improves Public Access and Internal Decision-Making
AI without source citations creates real liability. Learn how citation-backed AI brings traceable sources, version awareness, and audit-ready outputs to every institutional decision.
Data and AI Solutions Group
Marketing Group view
How to Perform a Successful Network Penetration Test: Comprehensive Guide for 2025
Learn how to perform a successful network penetration test to identify vulnerabilities, simulate real cyberattacks, and strengthen your organization’s network security.
Cybersecurity Solutions Group
Marketing Group view
What Is Penetration Testing? A 2026 Expert Guide
A 2026 expert guide to penetration testing for security leaders and IT teams seeking proactive defense, compliance, and stakeholder trust.
Cybersecurity Solutions Group
Marketing Group view
OT Ransomware Prevention: Practical Best Practices for Industrial Cybersecurity
Explore enterprise grade OT ransomware prevention strategies, including segmentation, identity control, threat informed detection, and resilient recovery design to protect industrial operations fro
Cybersecurity Solutions Group
Marketing Group view
10 Myths About OT/ICS Security That Put Your Business at Risk
Think your OT network is secure? Learn the 10 most dangerous myths about OT and ICS cybersecurity that leave industrial operations exposed to attacks.
Cybersecurity Solutions Group
Marketing Group view
OT Ransomware Risks and Response for Industrial Systems
Learn why OT environments face higher ransomware risk, how attackers gain access, and how effective detection and response reduce operational impact.
Cybersecurity Solutions Group
Marketing Group view
AI Risk Assessment: Risk Types, Best Practices & More
Explore AI risk types, essential assessment frameworks, and proven best practices to mitigate threats in AI deployment. Learn actionable strategies for secure AI systems today.
Cybersecurity Solutions Group
Marketing Group view
AI Risk Assessment: Everything You Need to Know
Learn essential processes, methodologies, risk types, regulatory requirements, and practical implementation strategies for safe AI deployment.
Cybersecurity Solutions Group
Marketing Group view
Whitepaper: Ransomware Threat Management
Ransomware continues to be a real threat to business operations across all industries, no organization is safe from this threat.
Laszlo S. Gonc
CISSP, First Senior Fellow, DivIHN Cybersecurity Center of Excellence view
Cybersecurity Incident Response Preparedness
An incident response framework provides a structure to support incident response operations. A framework typically provides guidance on what needs to be done, but not on how it is done.
Laszlo S. Gonc
CISSP, First Senior Fellow, DivIHN Cybersecurity Center of Excellence view
IoT Medical Device Cybersecurity
Healthcare data and medical devices would be aggressively targeted by ransomware attacks since early 2017 has proven to be true






